Structure variants calling and genotyping scripts for pan-Zea.
The general pipelines were graphically illustrated as:
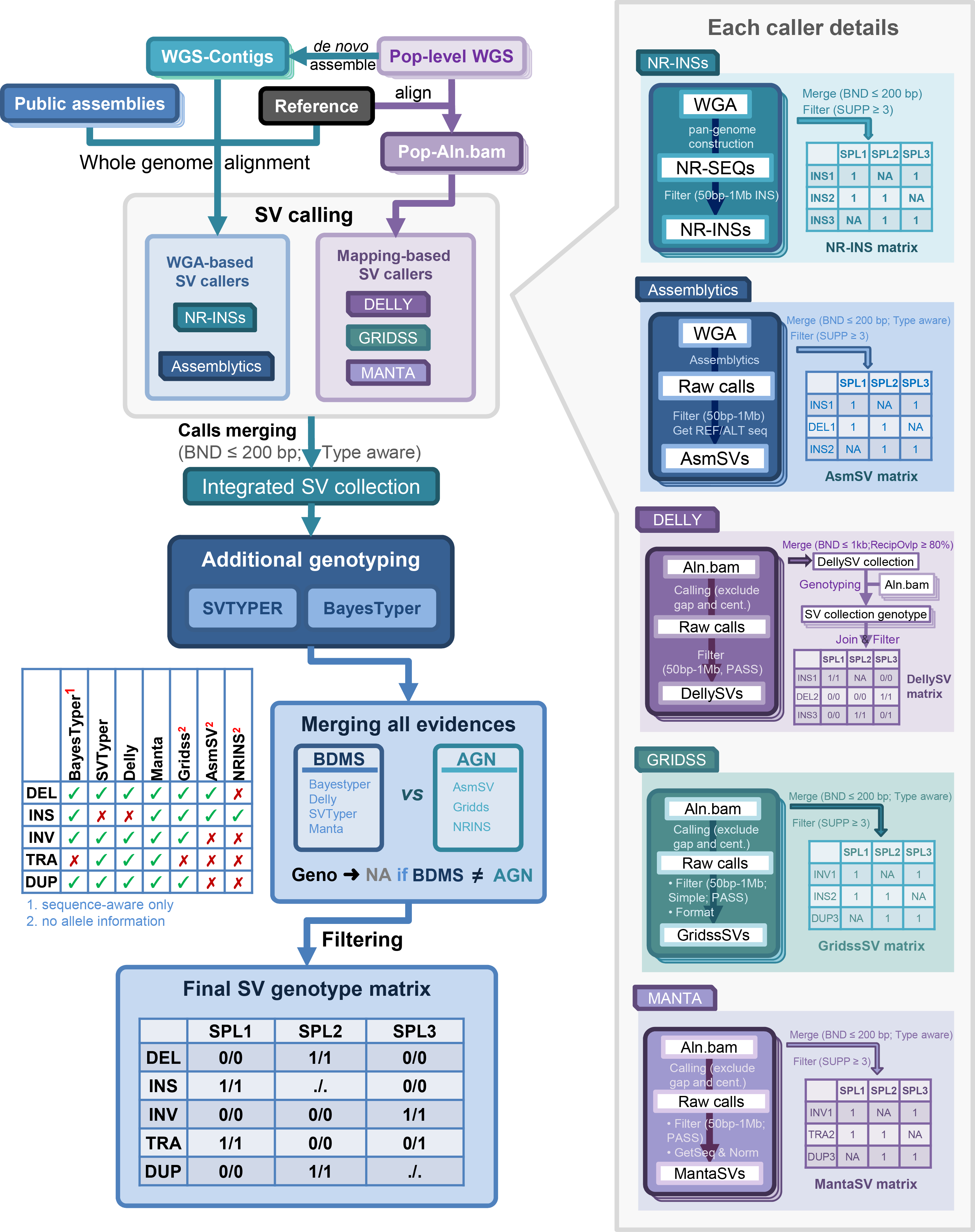
See more details in the pan-Zea genome research article.
- Linux, tested with v3.10.0-862.el7.x86_64 (Red Hat 4.8.5-28)
- Rscript, tested with v3.6.0
- perl 5, tested with v5.30.1
| Code_Dir | Description |
|---|---|
Call-Asm-Assemblytics/ |
custom scripts in Assemblytics based SV calling |
Call-Asm-NRINS/ |
custom scripts in parsing NR-INSs from pan-Zea_construct NR-SEQs |
Call-WGS-delly/ |
custom scripts in dealing with delly SV calls |
Call-WGS-gridss/ |
custom scripts in dealing with gridss SV calls |
Call-WGS-manta/ |
custom scripts in dealing with manta sv calls |
Call-ZZ-merge/ |
custom scripts to merge all callers' results |
Genotyping/ |
custom scripts in additional genotyping and merging results from all genotypers |
NOTE:
-
This repository did not contain the executables of each caller and the detail parameters used in each caller (which were already documented in the related research article)
-
Some custom scripts assumed the correct name format to parse the information, so we do not guarantee a bug-free running of these scripts on other projects.
-
Call-Asm-Assemblytics/Assemblytics-mdf: modified Assemblytics main script to expand the maximum size of SV to 1Mb. To make it work, just replace theAssemblyticsscript of the original Assemblytics repository with this script. -
Call-Asm-Assemblytics/run_asmSV.sh: given a WGA result between the query and ref genome in paf.gz format, output a VCF file of SVs. A wrapper of Assemblytics --> SURVIVOR------------------------------------------------------------ run modified assemblytics and SURVIVOR ------------------------------------------------------------ Dependence: Assemblytics: modified version that support SVs up to 1Mb [SURVIVOR](https://github.com/fritzsedlazeck/SURVIVOR) [paf2delta.py](https://gist.github.com/malonge/d347a0061eab77d9ebeb9181b7a9ff31) ------------------------------------------------------------ USAGE: bash $(basename $0) <pre> <paf.gz> pre: the output prefix, usually be the name of the individual paf.gz: path to the WGA result between the query genome and the ref genome, in paf format NOTE: Before running, make sure the "assemblytics_dir" in line 5 of this script was correctly set to the right path of your local Assemblytics installation ------------------------------------------------------------ -
Call-Asm-Assemblytics/paf2delta.py: convert paf format to delta format, forked from gist.github.com/malonge/paf2delta.py -
Call-Asm-Assemblytics/fmt_asmSV.sh: format vcf output ofCall-Asm-Assemblytics/run_asmSV.sh------------------------------------------------------------ A script for formatting asmSV.vcf IDs ------------------------------------------------------------ USAGE: $0 <input.vcf (result from run_asmSV.sh)> Parse input names to get [SP]: input names should be in format of "[SP]_asmSV_XXXX.vcf" ------------------------------------------------------------
-
Call-Asm-Assemblytics/get_asmSV_seq.sh: get the sequences of ref and alt allele of Assemblytics outputs------------------------------------------------------------ A script for getting the exact seq from Assemblytics results ------------------------------------------------------------ USAGE: $0 [options below] 1. ABS_PATH of vcf file of the Assemblytics results,named: PREFIX_asmSV_mft.vcf (converted by SURVIVOR and reformated by local script) 1. ABS_PATH of Raw Assemblytics results dir 2. ABS_PATH of REF_fasta 3. ABS_PATH of Query_fasta ------------------------------------------------------------
-
Call-Asm-NRINS/Filter_NRINS_and_GetSeq.sh: filter NR-SEQ records to get NR-INSs------------------------------------------------------------ Filter NRINS VCFs and get sequence: Keep only both end anchored to same chr records. Filter out ref or query region > 1 Mb. ------------------------------------------------------------ USAGE: bash $(basename $0) <NRINS.vcf> <Ref_seq.fa> <Query_seq.fa> <prefix> ------------------------------------------------------------ -
Call-Asm-NRINS/run_SRVIVOR_200.sh: Merge allNRINS vcfs using SURVIVOR with breakpoints interval cutoff of 200 bp and minimum SV len of 50 bp, then filter out records that had less than 3 population level supports -
Call-Asm-NRINS/filter_SURVIVOR.sh: Filter merged SURVIVOR VCF by population level supports------------------------------------------------------------ Filter survivor merge vcf files by support samples ------------------------------------------------------------ USAGE: filter_SURVIVOR.sh <survivor_merge.vcf> <supp cut-off> <Prefix> Prefix: for non-sample out vcfs, usually use Method, eg: Manta ------------------------------------------------------------
-
Call-WGS-delly/01_batch_delly_raw.sh: script to generate PBS jobs for delly raw SV calling of each individual -
Call-WGS-delly/02_delly_merge.sh: PBS job of merging all delly calls -
Call-WGS-delly/03_batch_delly_geno.sh: script to generate PBS jobs for delly SV genotyping of each individual based on merged delly calls -
Call-WGS-delly/04_delly_filter.sh: PBS job of filtering population level delly genotyping result
-
Call-WGS-gridss/gst_gridss_anno_filt_bed.R: filtering the raw gridss vcf output for each individual, by keeping only simple (INS, INV, DEL, DUP) records that with quality tag "PASS". Modified from the official gridss simple-event-annotation.R. -
Call-WGS-gridss/00_Get_Gridss_seq_vcf.sh: get ref alt sequences from Gridss simple SV outputs and Gridss raw SV outputs
manta call flowchart:
s=>start: Each bam
A=>operation: Raw call
B=>operation: Filter and Get allele
con=>condition: Seq-aware ?
C=>operation: Seq-aware SVs
D=>operation: Other SVs
E=>operation: Norm allele
F=>operation: Combine and format
G=>operation: Merge and Pop-filter
H=>operation: get raw manta geno
e=>end: Final Manta SV matrix
s->A->B->con
con(yes)->C->E->F
con(no)->D->F->G->H->e
-
Call-WGS-manta/01_batch_manta.sh: script to generate the PBS jobs for each individual's manta raw SV calling. -
Call-WGS-manta/02_batch_filter.sh: script to generate the PBS jobs to filter the raw calls and get the sequence-aware records of each individual's manta raw SV calls. Bayestyper should be installed and assigned to$PATHto make this script work. -
Call-WGS-manta/03_batch_norm.sh: script to generate the PBS jobs for normalizing the sequence-aware SVs with bcftools. -
Call-WGS-manta/04_fmt_manta_with_seq.sh: combine sequence-aware calls with the other manta SV calls, remove duplications and reformat names.------------------------------------------------------------ combine raw manta with convert allele out, rmdup and rename ------------------------------------------------------------ Dependency: csvtk bcftools ------------------------------------------------------------ USAGE: 04_fmt_manta_with_seq.sh <raw_manta.vcf> <convert_allele.vcf.gz> <cpus> ------------------------------------------------------------
-
Call-WGS-manta/05_from_sur_to_raw_manta_geno.sh: script to replace the SURVIVOR genotypes with the raw manta genotypes for that item.------------------------------------------------------------ Convert Genotype in SURVIVOR merged manta result back into there raw genotype format ------------------------------------------------------------ Dependence: csvtk parallel ------------------------------------------------------------ USAGE: bash $(basename $0) <survivor_merged_vcf> <raw_each_sample_vcf_list> <output> <threads> list format: <sample_ID> <File_Path> ------------------------------------------------------------
The SV calls of each caller were merged if they were the same SV type and their breakpoints were within 200 bp using SURVIVOR version 1.0.6.
The representations of the merged calls were set using the Genotyping/ZZ_Merge-all/PANZ_SV_Geno_merge.sh:
------------------------------------------------------------
USAGE:
bash ParsePANZ_MergeSV.sh <input: 00_Merge_all.vcf>
------------------------------------------------------------
A script to get represent record for Merged All SV for PANZ
rename the SV, get represent ref-alt seq, and generate these
outputs:
PANZ_SV_Reformat.vcf
PANZ_SV_SVtyper.vcf
PANZ_SV_Delly.vcf
PANZ_SV_Bayestyper.vcf
PANZ_SV_ID_each_call.tsv
Record Keep Priority:
1BSpSV > 2AsmREF > 3AsmAMP > 4NRINS > 5Gridss > 6Manta > 7Delly
Rename rules:
PZ00001aSVnum[SVTYPE]Ref_Chr1#Posi1#Chr2#Pos2
Note: input vcf should been merged according to the priority order above.
(The script parsed the SUPP_VEC=010101 information without checking the order.)
------------------------------------------------------------Genotyping/SVTyper/batch_svtyper_lsf.sh: script to batch generate the LSF jobs of SVTyper genotyping of the merged SVs for each individual.
The genotyped vcf outputs were joined using bcftools merge with default options.
-
Genotyping/Bayestyper/get_rand_snpindel_SVs.sh: script to randomly select ~1 million (1/10 of the total SNPs and INDELs) SNP and INDELs for SV genotyping. -
Genotyping/Bayestyper/00_run_bys.sh: a wrapper of Bayestyper genotyping pipeline.------------------------------------------------------------ Run bayestyper cluster and genotype ------------------------------------------------------------ USAGE: bash get_rand_snpindel_SVs.sh [OPTIONs] OPT: 1. simple_ist 2. vcf_file 3. ref_fasta 4. decoy_fasta 5. threads ------------------------------------------------------------
Genotyping/ZZ_Merge-all/PANZ_SV_Geno_merge.sh: script to merge all the genotype evidences to get the consistent genotype matrix
------------------------------------------------------------
Merging all SV geno evidences to get final genotype
------------------------------------------------------------
USAGE:
bash PANZ_SV_Geno_merge.sh [OPTIONS]
OPTIONS: ([R]:required [O]:optional)
-h, --help show help and exit.
-v, --vcf <str> [R] Raw SV records vcf file
-l, --list <str> [R] List of sample ID name map, will use the name and the order of this file
to generate the final result, so make sure the sample names are unique.
Format (Tab-sep-table):
<ID> <Sample Name>
Eg.:
Q417 SK
Q441 B73
--genorate <0-1> [O] Genotype rate cutoff for filtering (Default: 0.1)
--maf <0-1> [O] Minus allel frequence cutoff for filtering (Default: 0.01)
-b, --bayestyper <str> [R] bayestyper genotype matrix
-d, --delly <str> [R] delly genotype matrix
-s, --svtyper <str> [R] svtyper genotype matrix
-m, --manta <str> [R] manta genotype matrix
-a, --asmsv <str> [R] AsmSV genotype matrix
-g, --gridss <str> [R] Gridss genotype matrix
-n, --nrins <str> [R] NR-INS genotype matrix
-o, --output <str> [O] Output file name (default: 00_PANZ_merged_geno.matrix)
-t, --threads <num> [O] set threads (default 2)
-w, --weight <str> [O] comma seperated integer weight value for setting the genotypes, format:
bayestyper_w,delly_w,manta_w,svtyper_w
(Default: \"2,1,1,1\")
-c, --cutoff <0-1> [O] Cut-off value to set final genotype (Default 0.5)
------------------------------------------------------------If you use this pipeline in your work, or you would like to know more details, please refer to:
Gui, S., Wei, W., Jiang, C. et al. A pan-Zea genome map for enhancing maize improvement. Genome Biol 23, 178 (2022). https://doi.org/10.1186/s13059-022-02742-7